
Inherited Retinal Diseases
Clinical features and molecular mechanisms of RP1L1 variants causing occult macular dystrophy
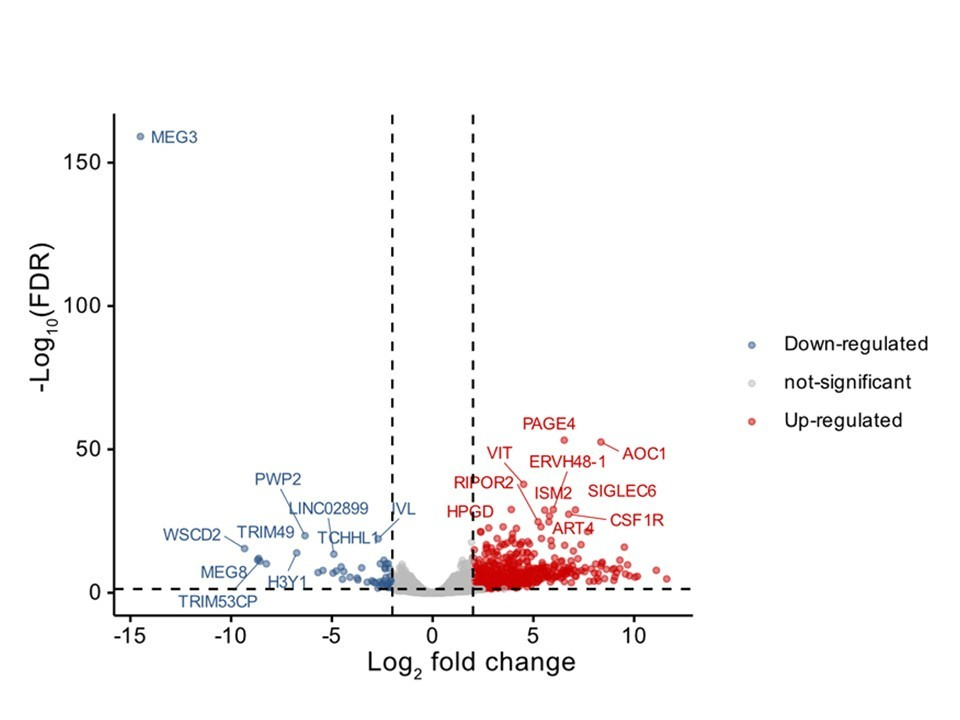
Occult macular dystrophy (OMD) is an inherited retinopathy characterized by progressive bilateral vision loss despite normal findings on fundoscopic examination, fluorescein angiography, and full-field electroretinography. Its pathogenesis remains unknown, and no treatments are available. Here, we performed whole-exome sequencing on 133 samples from 78 OMD pedigrees to identify pathogenic variants, using filters for minor allele frequency, function prediction, and retinal expression. We identified the RP1L1 c.133C>T, p.Arg45Trp (R45W) mutation as the sole pathogenic variant in two families with dominantly inherited OMD. Additionally, we discovered five other potentially pathogenic RP1L1 variants. Together, these six variants accounted for 33.33% of pedigrees, with R45W being the most prevalent at 16.6%. The R45W mutation correlated with earlier onset, more severe clinical phenotypes, and abnormal intracellular localization rather than altered expression levels. R45W disrupted the intracellular localization of RP1L1 and RP1, compromising cell viability. In induced photoreceptor-like cells derived from OMD patients carrying R45W, we observed downregulation of the long noncoding RNA MEG3 and the PI3K/Akt pathway, alongside upregulation of extracellular matrix organization. These findings validate the etiologic role of RP1L1 and offer insights into the pathogenesis of OMD, thereby facilitating future research and therapeutic development.
Pan Y, Iejima D, Yoshitake K, Tsunoda K, Iwata T.
HGG Advances 2025 May 31, 100461
Compound heterozygous mutations in a mouse model of Leber congenital amaurosis reveal the role of CCT2 in photoreceptor maintenance.

TRiC/CCT is a chaperonin complex required for the folding of cytoplasmic proteins. Although mutations in each subunit of TRiC/CCT are associated with various human neurodegenerative diseases, their impact in mammalian models has not yet been examined. A compound heterozygous mutation in CCT2 (p.[Thr400Pro]; p.[Arg516His]) is causal for Leber congenital amaurosis. Here, we generate mice carrying each mutation and show that Arg516His (R516H) homozygosity causes photoreceptor degeneration accompanied by a significant depletion of TRiC/CCT substrate proteins in the retina. In contrast, Thr400Pro (T400P) homozygosity results in embryonic lethality, and the compound heterozygous mutant (T400P/R516H) mouse showed aberrant cone cell lamination and died 2 weeks after birth. Finally, CCDC181 is identified as a interacting protein for CCTβ protein, and its localization to photoreceptor connecting cilia is compromised in the mutant mouse. Our results demonstrate the distinct impact of each mutation in vivo and suggest a requirement for CCTβ in ciliary maintenance.
Suga A, Minegishi Y, Yamamoto M, Ueda K, Iwata T.
Commun Biol. 2024 Jun 3;7(1):676. doi: 10.1038/s42003-024-06384-2. PMID: 38830954
A homozygous structural variant of RPGRIP1 is frequently associated with achromatopsia in Japanese patients with IRD

Achromatopsia (ACHM) is an early-onset cone dysfunction caused by 5 genes with cone-specific functions (CNGA3, CNGB3, GNAT2, PDE6C, and PDE6H) and by ATF6, a transcription factor with ubiquitous expression. To improve the relatively low variant detection ratio in these genes in a cohort of exome-sequenced Japanese patients with inherited retinal diseases (IRD), we performed genome sequencing to detect structural variants and intronic variants in patients with ACHM. Genome sequencing of 10 ACHM pedigrees was performed after exome sequencing. Structural, non-coding, and coding variants were filtered based on segregation between the affected and unaffected in each pedigree. Variant frequency and predicted damage scores were considered in identifying pathogenic variants. A homozygous deletion involving exon 18 of RPGRIP1 was detected in 5 of 10 ACHM probands, and variant inheritance from each parent was confirmed. This deletion was relatively frequent (minor allele frequency = 0.0023) in the Japanese population but was only homozygous in patients with ACHM among the 199 Japanese IRD probands analyzed by the same genome sequencing pipeline.
The deletion involving exon 18 of RPGRIP1 is a prevalent cause of ACHM in
Japanese patients and contributes to the wide spectrum of RPGRIP1-associated IRD phenotypes, from Leber congenital amaurosis to ACHM.
Suga A, Mizobuchi K, Inooka T, Yoshitake K, Minematsu N, Tsunoka K, Kuniyoshi K, Kawai Y, Omae Y, Tokunaga K, NCBN Controls WGS Consortium, Hayashi T, Ueno S, Iwata T.
Genetics in Medicine Open, 2024 March 2(15):101843. DOI:10.1016/j.gimo.2024.101843
Genetic characterization of 1,210 Japanese pedigrees with inherited retinal diseases by whole-exome sequencing
 Inherited retinal diseases (IRDs) comprise a phenotypically and genetically heterogeneous group of ocular disorders that cause visual loss via progressive retinal degeneration. Here we report the genetic characterization of 1,210 IRD pedigrees enrolled through the Japan Eye Genetic Consortium and analyzed by whole exome sequencing. The most common phenotype was retinitis pigmentosa (RP, 43%), followed by macular dystrophy/cone- or cone-rod dystrophy (MD/CORD, 13%). In total, 67 causal genes were identified in 37% (448/1210) of the pedigrees. The first and second most frequently mutated genes were EYS and RP1, associated primarily with autosomal recessive (ar) RP, and RP and arMD/CORD, respectively. Examinations of variant frequency in total and by phenotype showed high accountability of a frequent EYS missense variant (c.2528G>A). In addition to the two known EYS founder mutations (c.4957dupA and c.8805C>G) of arRP, we observed a frequent RP1 variant (c.5797C>T) in patients with arMD/CORD. This article is protected by copyright. All rights reserved.
Inherited retinal diseases (IRDs) comprise a phenotypically and genetically heterogeneous group of ocular disorders that cause visual loss via progressive retinal degeneration. Here we report the genetic characterization of 1,210 IRD pedigrees enrolled through the Japan Eye Genetic Consortium and analyzed by whole exome sequencing. The most common phenotype was retinitis pigmentosa (RP, 43%), followed by macular dystrophy/cone- or cone-rod dystrophy (MD/CORD, 13%). In total, 67 causal genes were identified in 37% (448/1210) of the pedigrees. The first and second most frequently mutated genes were EYS and RP1, associated primarily with autosomal recessive (ar) RP, and RP and arMD/CORD, respectively. Examinations of variant frequency in total and by phenotype showed high accountability of a frequent EYS missense variant (c.2528G>A). In addition to the two known EYS founder mutations (c.4957dupA and c.8805C>G) of arRP, we observed a frequent RP1 variant (c.5797C>T) in patients with arMD/CORD. This article is protected by copyright. All rights reserved.
Suga A, Yoshitake K, Minematsu N, Tsunoda K, Fujinami K, Miyake Y, Kuniyoshi K, Hayashi T, Mizobuchi K, Ueno S, Terasaki H, Kominami T, Nao-I N, Mawatari G, Mizota A, Shinoda K, Kondo M, Kato K, Sekiryu T, Nakamura M, Kusuhara S, Yamamoto H, Yamamoto S, Mochizuki K, Kondo H, Matsushita I, Kameya S, Fukuchi T, Hatase T, Horiguchi M, Shimada Y, Tanikawa A, Yamamoto S, Miura G, Ito N, Murakami A, Fujimaki T, Hotta Y, Tanaka K, Iwata T.
Hum Mutat. 2022 Oct 25. doi: 10.1002/humu.24492. Online ahead of print.PMID: 36284460
LRRTM4-C538Y novel gene mutation is associated with hereditary macular degeneration with novel dysfunction of ON-type bipolar cells
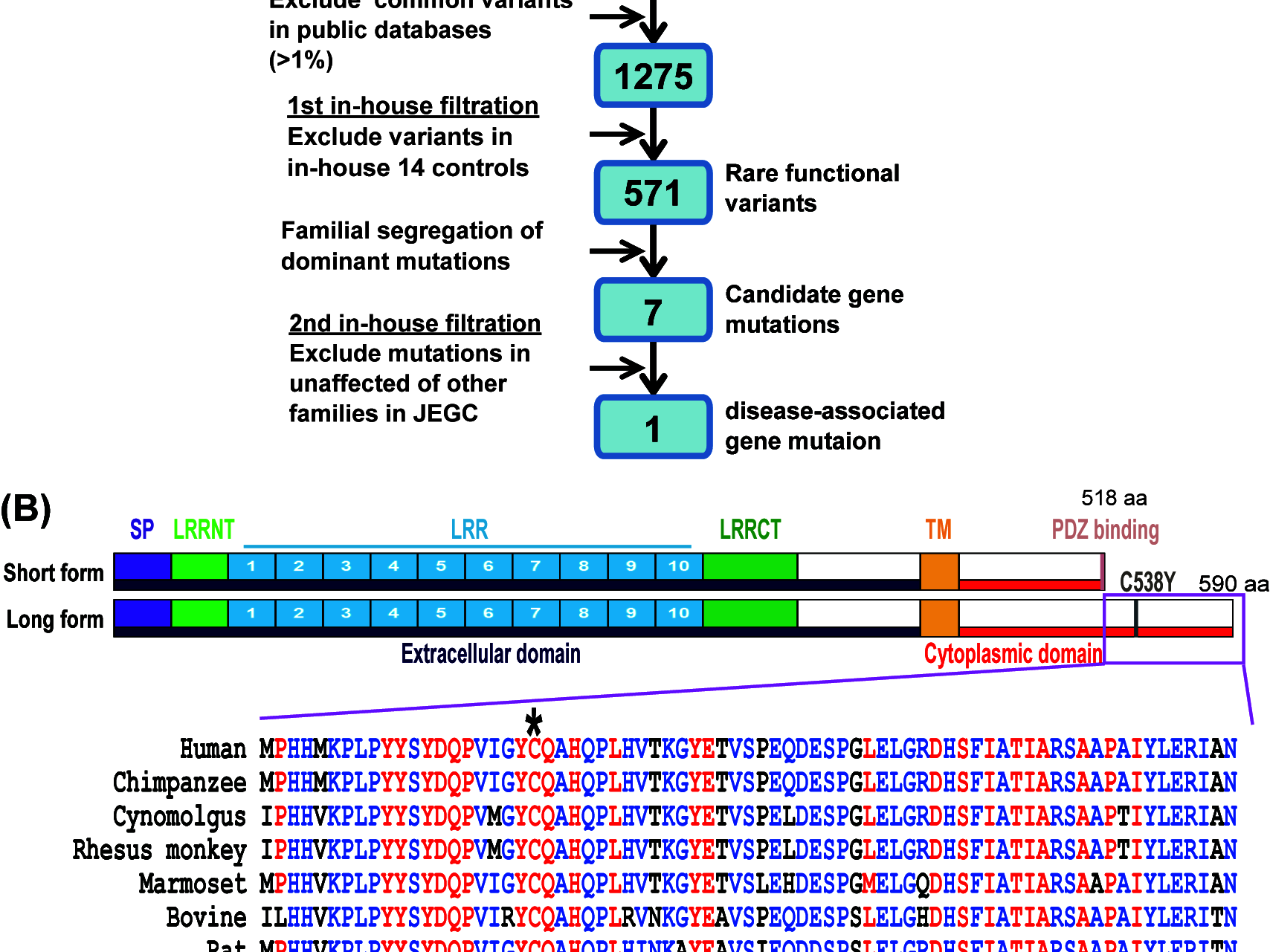 The macula is a unique structure in higher primates, where cone and rod photoreceptors show highest density in the fovea and the surrounding area, respectively. The hereditary macular dystrophies represent a heterozygous group of rare disorders characterized by central visual loss and atrophy of the macula and surrounding retina. Here we report an atypical absence of ON-type bipolar cell response in a Japanese patient with autosomal dominant macular dystrophy (adMD). To identify a causal genetic mutation for the adMD, we performed whole-exome sequencing (WES) on four affected and four-non affected members of the family for three generations, and identified a novel p.C538Y mutation in a post-synaptic gene, LRRTM4. WES analysis revealed seven rare genetic variations in patients. We further referred to our in-house WES data from 1360 families with inherited retinal diseases, and found that only p.C538Y mutation in LRRTM4 was associated with adMD-affected patients. Combinatorial filtration using public database of single-nucleotide polymorphism frequency and genotype-phenotype annotated database identified novel mutation in atypical adMD.
The macula is a unique structure in higher primates, where cone and rod photoreceptors show highest density in the fovea and the surrounding area, respectively. The hereditary macular dystrophies represent a heterozygous group of rare disorders characterized by central visual loss and atrophy of the macula and surrounding retina. Here we report an atypical absence of ON-type bipolar cell response in a Japanese patient with autosomal dominant macular dystrophy (adMD). To identify a causal genetic mutation for the adMD, we performed whole-exome sequencing (WES) on four affected and four-non affected members of the family for three generations, and identified a novel p.C538Y mutation in a post-synaptic gene, LRRTM4. WES analysis revealed seven rare genetic variations in patients. We further referred to our in-house WES data from 1360 families with inherited retinal diseases, and found that only p.C538Y mutation in LRRTM4 was associated with adMD-affected patients. Combinatorial filtration using public database of single-nucleotide polymorphism frequency and genotype-phenotype annotated database identified novel mutation in atypical adMD.
Kawamura Y, Suga A, Fujimaki T, Yoshitake K, Tsunoda K, Murakami A, Iwata T.
J Hum Genet. 2018 Aug;63(8):893-900. doi: 10.1038/s10038-018-0465-4. Epub 2018 May 14. PMID: 29760528
CCT2 Mutations Evoke Leber Congenital Amaurosis due to Chaperone Complex Instability
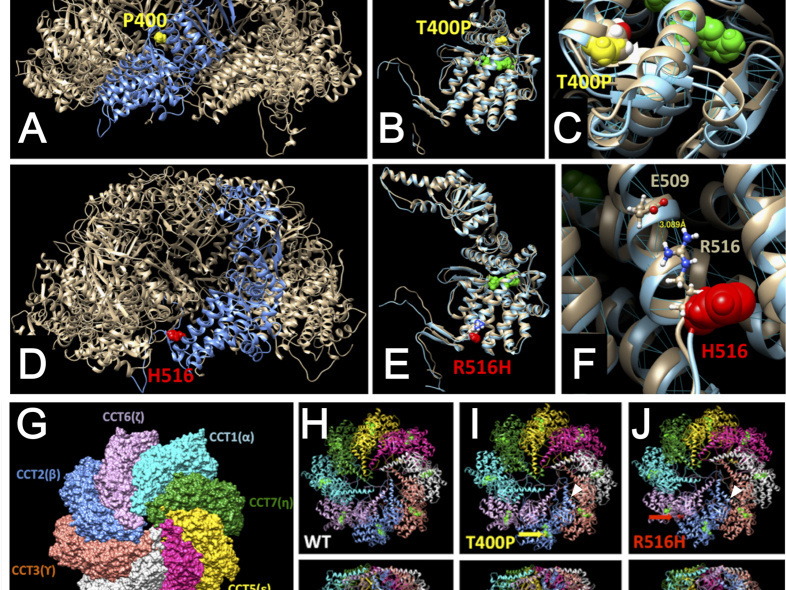 Leber congenital amaurosis (LCA) is a hereditary early-onset retinal dystrophy that is accompanied by severe macular degeneration. In this study, novel compound heterozygous mutations were identified as LCA-causative in chaperonin-containing TCP-1, subunit 2 (CCT2), a gene that encodes the molecular chaperone protein, CCTβ. The zebrafish mutants of CCTβ are known to exhibit the eye phenotype while its mutation and association with human disease have been unknown. The CCT proteins (CCT α-θ) forms ring complex for its chaperon function. The LCA mutants of CCTβ, T400P and R516H, are biochemically instable and the affinity for the adjacent subunit, CCTγ, was affected distinctly in both mutants. The patient-derived induced pluripotent stem cells (iPSCs), carrying these CCTβ mutants, were less proliferative than the control iPSCs. Decreased proliferation under Cct2 knockdown in 661W cells was significantly rescued by wild-type CCTβ expression. However, the expression of T400P and R516H didn't exhibit the significant effect. In mouse retina, both CCTβ and CCTγ are expressed in the retinal ganglion cells and connecting cilium of photoreceptor cells. The Cct2 knockdown decreased its major client protein, transducing β1 (Gβ1). Here we report the novel LCA mutations in CCTβ and the impact of chaperon disability by these mutations in cellular biology.
Leber congenital amaurosis (LCA) is a hereditary early-onset retinal dystrophy that is accompanied by severe macular degeneration. In this study, novel compound heterozygous mutations were identified as LCA-causative in chaperonin-containing TCP-1, subunit 2 (CCT2), a gene that encodes the molecular chaperone protein, CCTβ. The zebrafish mutants of CCTβ are known to exhibit the eye phenotype while its mutation and association with human disease have been unknown. The CCT proteins (CCT α-θ) forms ring complex for its chaperon function. The LCA mutants of CCTβ, T400P and R516H, are biochemically instable and the affinity for the adjacent subunit, CCTγ, was affected distinctly in both mutants. The patient-derived induced pluripotent stem cells (iPSCs), carrying these CCTβ mutants, were less proliferative than the control iPSCs. Decreased proliferation under Cct2 knockdown in 661W cells was significantly rescued by wild-type CCTβ expression. However, the expression of T400P and R516H didn't exhibit the significant effect. In mouse retina, both CCTβ and CCTγ are expressed in the retinal ganglion cells and connecting cilium of photoreceptor cells. The Cct2 knockdown decreased its major client protein, transducing β1 (Gβ1). Here we report the novel LCA mutations in CCTβ and the impact of chaperon disability by these mutations in cellular biology.
Minegishi Y, Sheng X, Yoshitake K, Sergeev Y, Iejima D, Shibagaki Y, Monma N, Ikeo K, Furuno M, Zhuang W, Liu Y, Rong W, Hattori S, Iwata T.
Sci Rep. 2016 Sep 20;6:33742. doi: 10.1038/srep33742. PMID: 27645772
Identification of novel mutations in the LRR-cap domain of C21orf2 in Japanese patients with retinitis pigmentosa and cone-rod dystrophy
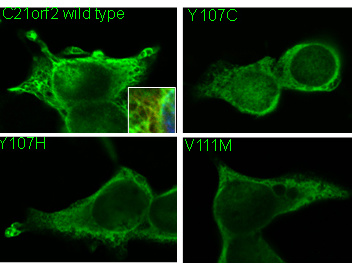 Cytoplasmic localization of Flag-tagged C21orf2 wild type and mutant proteins. Insert shows overlapping Flag (green) and Tubulin (red) immunostaining.Inherited retinal dystrophy is a rare disease, but has a critical impact on the patient’s quality of life, in that fundamental medical treatments are almost not available until recently. Latest advances of gene therapy technologies and stem cell-based drug screenings are changing the situation, yet, it is still under investigation how the variety of genetic mutations causes the disease, and, further, causal genes/mutations are not identified in up to 30% of the cases.
Cytoplasmic localization of Flag-tagged C21orf2 wild type and mutant proteins. Insert shows overlapping Flag (green) and Tubulin (red) immunostaining.Inherited retinal dystrophy is a rare disease, but has a critical impact on the patient’s quality of life, in that fundamental medical treatments are almost not available until recently. Latest advances of gene therapy technologies and stem cell-based drug screenings are changing the situation, yet, it is still under investigation how the variety of genetic mutations causes the disease, and, further, causal genes/mutations are not identified in up to 30% of the cases.
To identify novel causal genes and mutations of inherited retinal diseases in Japanese population, we have been collecting 872 families with patient(s) and their healthy siblings and/or parents, and working on Whole Exome Sequencing (WES) analysis on 594 families.
One of the novel mutations found in this study, we reported mutations in a ciliary gene, C21orf2, from the patients affected by autosomal recessive retinitis pigmentosa (arRP) and autosomal recessive cone-rod dystrophy (arCRD), respectively.
RP is the most common inherited retinal dystrophy with the prevalence of 1 in 4,000 worldwide. The disease onset and progression of RP is highly variable. However, many patients fall into night blindness followed by the loss of mid-peripheral visual field, which are caused by the rod photoreceptor degeneration. CRD is a further rare disease with the prevalence of 1 in 40,000. Patients typically have photoaversion, decreased visual acuity and loss of central visual field, reflecting the degeneration of cone photoreceptors. RP and CRD are genetically highly variable: RP and CRD can be inherited in any type of hereditary manner; autosomal dominant, autosomal recessive, and X-linked. Further, RP and CRD have at least 80 and 30 causal genes, respectively, with those numbers increasing. In addition, RP and CRD can appear as an associating symptom of syndromic diseases, such as Jubert syndrome, Bardet-Biedl syndrome, Jeune syndrome, and Usher syndrome type 2.
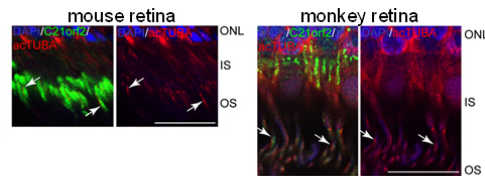 Localization of C21orf2 protein (green) in the photoreceptor connecting cilium indicated by the acetylated Tubulin (red). Mutations in C21orf2 were identified by family-based genetic segregations. The arRP patients had compound heterozygous mutation (p. Y107H, p. V111M), and the arCRD patients had homozygous mutation (p. Y107C). All these mutated amino acids were conserved from human to a unicellular organism, chlamydomonas, suggesting their importance in the protein function. In fact, in vitro studies showed that the mutated C21orf2 proteins expressed at lower levels, and diffusely distributed in the cytoplasm compared to the wild type C21orf2 protein co-localizing to the Tubulin. C21orf2 protein localized to the primary cilia of rodent and primate photoreceptors in vivo. Since C21orf2 was required for ciliogenesis, our data suggested that reduced levels of functional C21orf2 induced photoreceptor degradation through abnormal cilia formation, leading to arRP or arCRD in the retina. C21orf2 was also identified in atypical retinal dystrophy patients and also in the patients with syndromic skeletal dystrophy. Different effects of each mutation and genetic backgrounds may result in the symptom isolated to the eye or syndromic diseases.
Localization of C21orf2 protein (green) in the photoreceptor connecting cilium indicated by the acetylated Tubulin (red). Mutations in C21orf2 were identified by family-based genetic segregations. The arRP patients had compound heterozygous mutation (p. Y107H, p. V111M), and the arCRD patients had homozygous mutation (p. Y107C). All these mutated amino acids were conserved from human to a unicellular organism, chlamydomonas, suggesting their importance in the protein function. In fact, in vitro studies showed that the mutated C21orf2 proteins expressed at lower levels, and diffusely distributed in the cytoplasm compared to the wild type C21orf2 protein co-localizing to the Tubulin. C21orf2 protein localized to the primary cilia of rodent and primate photoreceptors in vivo. Since C21orf2 was required for ciliogenesis, our data suggested that reduced levels of functional C21orf2 induced photoreceptor degradation through abnormal cilia formation, leading to arRP or arCRD in the retina. C21orf2 was also identified in atypical retinal dystrophy patients and also in the patients with syndromic skeletal dystrophy. Different effects of each mutation and genetic backgrounds may result in the symptom isolated to the eye or syndromic diseases.
Suga A, Mizota A, Kato M, Kuniyoshi K, Yoshitake K, Sultan W, Yamazaki M, Shimomura Y, Ikeo K, Tsunoda K, Iwata T.
Invest Ophthalmol Vis Sci 2016;57:4255-4263
Dominant mutations in RP1L1 gene are responsible for occult macular dystrophy (Miyake's Disease)
 Occult Macular Dystrophy (OMD) is an autosomal dominant or sporadic form of macular dystrophy characterized by progressive decrease of visual acuity due to macular dysfunction. The disorder was called 'occult' by the fact that the macular dysfunction of this disease is hidden by a normal fundus appearance. Typical OMD, as described by Miyake et al is characterized by central cone dysfunction and in some cases rod dysfunction, leading to a loss of vision despite normal ophthalmoscopic appearance, normal fluorescein angiography, and normal full-field electroretinogram (ERGs); however, the amplitudes of the focal macular ERGs and multifocal ERGs are significantly reduced indicating dysfunction of the central retina. OMD is known for its broad range of disease onset from 6 to 81 years. Brockhurst et al have reported onset of four out of eight OMD patients at over 65 years and similar findings have also been observed in earlier cases.
Occult Macular Dystrophy (OMD) is an autosomal dominant or sporadic form of macular dystrophy characterized by progressive decrease of visual acuity due to macular dysfunction. The disorder was called 'occult' by the fact that the macular dysfunction of this disease is hidden by a normal fundus appearance. Typical OMD, as described by Miyake et al is characterized by central cone dysfunction and in some cases rod dysfunction, leading to a loss of vision despite normal ophthalmoscopic appearance, normal fluorescein angiography, and normal full-field electroretinogram (ERGs); however, the amplitudes of the focal macular ERGs and multifocal ERGs are significantly reduced indicating dysfunction of the central retina. OMD is known for its broad range of disease onset from 6 to 81 years. Brockhurst et al have reported onset of four out of eight OMD patients at over 65 years and similar findings have also been observed in earlier cases.
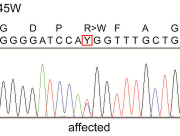 Linkage analysis of two OMD families collected by Tsunoda (NISO), Usui (Niigata Univ.), and Nakamura (Nakamura Eye Clinic) was performed by Fukuda and Tsuji (Tokyo Univ.) using SNP High Throughput Linkage analysis system (SNP HiTLink) localizing the disease locus to chromosome 8p22-23. Akahori (NISO) selected five candidate genes in the region, and found an amino acid substitution of Arg45Trp in retinitis pigmentosa 1-like 1 (RP1L1) gene was found in three OMD families and Trp960Arg in a remaining OMD family. These two mutations were detected in all affected individuals but none of the 876 controls. Immunohistochemistry of RP1L1 in the retina section of cynomolgus monkey revealed expression in the rod and cone photoreceptor, supporting the role of RP1L1 in the photoreceptors that, when disrupted by mutation, leads to OMD. Identification of RP1L1 as causative gene for OMD has potentially broader implications for understanding the differential cone photoreceptor functions in the fovea and the peripheral retina.
Linkage analysis of two OMD families collected by Tsunoda (NISO), Usui (Niigata Univ.), and Nakamura (Nakamura Eye Clinic) was performed by Fukuda and Tsuji (Tokyo Univ.) using SNP High Throughput Linkage analysis system (SNP HiTLink) localizing the disease locus to chromosome 8p22-23. Akahori (NISO) selected five candidate genes in the region, and found an amino acid substitution of Arg45Trp in retinitis pigmentosa 1-like 1 (RP1L1) gene was found in three OMD families and Trp960Arg in a remaining OMD family. These two mutations were detected in all affected individuals but none of the 876 controls. Immunohistochemistry of RP1L1 in the retina section of cynomolgus monkey revealed expression in the rod and cone photoreceptor, supporting the role of RP1L1 in the photoreceptors that, when disrupted by mutation, leads to OMD. Identification of RP1L1 as causative gene for OMD has potentially broader implications for understanding the differential cone photoreceptor functions in the fovea and the peripheral retina.
Akahori M, Tsunoda K, Miyake Y, Fukuda Y, Ishiura H, Tsuji S, Hatase T, Nakamua M, Ohde H, Itabashi T, Okamoto H, Takada Y, and Iwata T.
Am J Hum Genet 87:424-429 (2010)
